Research
I use computational and experimental approaches to study the ecology and evolution of bacteria, with a special interest in bacterial diversity, comparative genomics, and obligate slow-growing species. Below are some projects I've worked on, described from my perspective.
Complications stemming from horizontal gene transfer
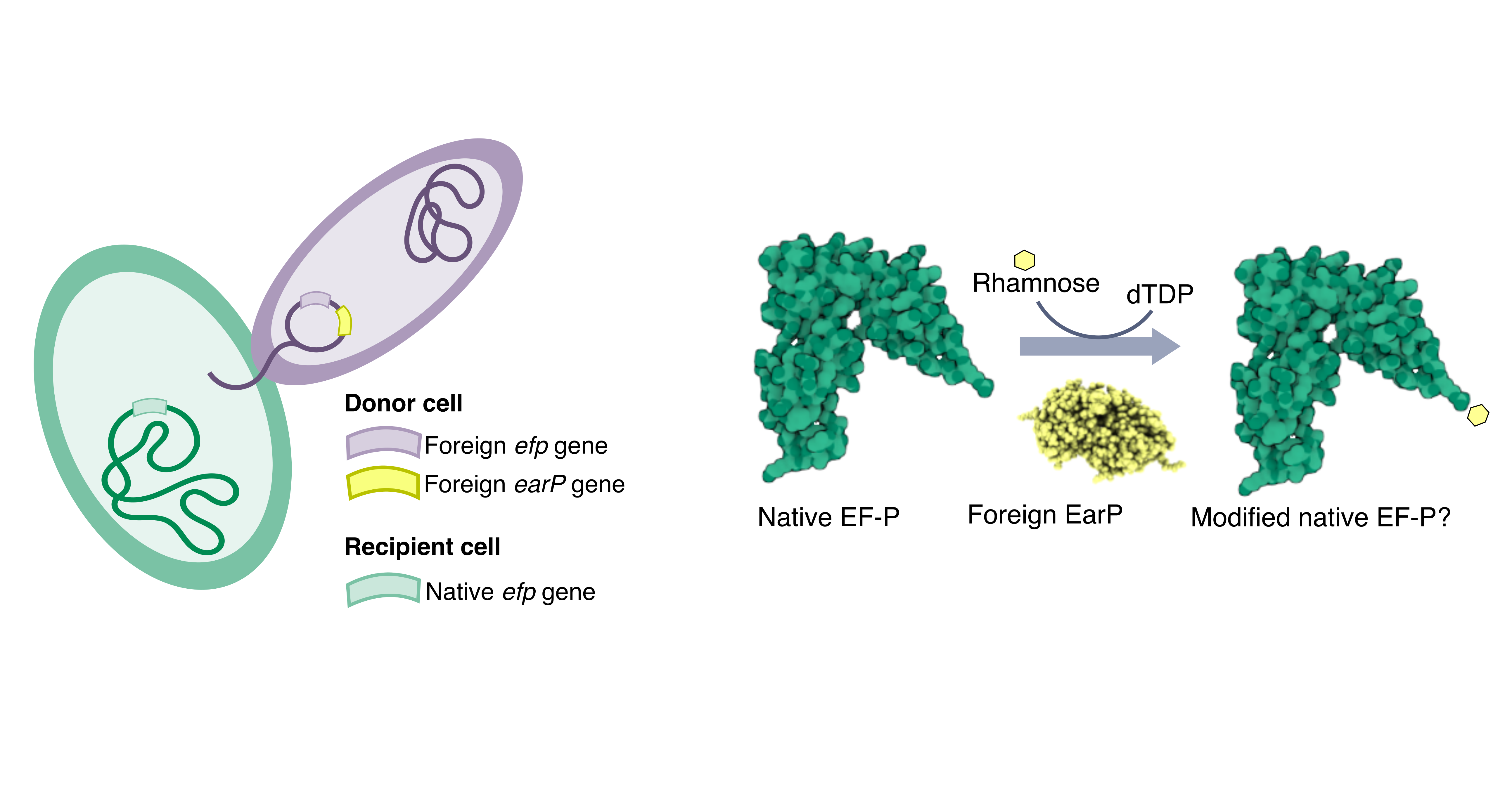
Horizontal transfer of an essential elongation factor is connected to loss of its target sequences This project centered around a genomic mystery. Elongation factor P (EF-P) is an essential translation factor which addresses a problem universal to all life: ribosomal stalling at polyproline motifs. (I can recommend this review on the subject, for further reading :)) I found that the horizontal transfer of new EF-P types is consistently associated with the loss of highly conserved polyproline motifs and the proteins that contain them. These patterns suggest that introducing a new EF-P system leads to a global disruption in polyproline translation. I hypothesized that this could stem from incompatibilities between horizontally transferred EF-P modification machinery and the native EF-P.
Horizontal transfer of proteins associated with EF-P can disrupt polyproline synthesis in a new host Here, I used heterologous expression, mass-spectrometry, and luminescent reporters to show that foreign post-translational modification machinery can modify non-target EF-P, impairing their function. Furthermore, we showed that this impact is not universal. While the EF-P from the Thermotogota Mesotoga prima seemed to be impaired by a misplaced modification, the EF-P subtype EfpL in E. coli was not affected by the same modification. This work highlighted that impacts of horizontally transferred genes vary based on cell background, leading to impairment in some, and possibilities for further adaption in others.
Connections between ribosomal efficiency and bacterial growth rates
Slow-growing bacteria encode more sequences which stall ribosomes Sequentially encoded prolines, due to their rigid structure, cause ribosomal stalling and disrupt translation. Fast-growing bacterial species, under strong pressure to optimize protein expression, are sensitive to such disruptions. Using computational analysis across 1000s of bacterial genomes, I found that fast-growing species encode fewer polyproline motifs. These motifs were enriched in signaling proteins tied to complex, multicellular lifestyles, suggesting growth optimization may limit evolutionary trajectories.
Extra copies of EF-P are associated with higer growth rates I also discovered a link between Gammaproteobacteria encoding a specialized EF-P variant (EfpL) and elevated growth rates. EF-P, a conserved translation factor, reduces ribosomal stalling at polyproline motifs. These analyses supported EfpL’s role as an evolutionary driver for enhanced bacterial growth.
Shedding light on uncultivated soil microbial communities
Genome streamlining in soil, Candidatus Udaeobacter copiosus. I led the analysis of one of the first metagenomically assembled genomes of an uncultivated soil bacterium, Candidatus Udaeobacter copiosus. Ca. Udaeobacter copiosus is a globally abundant, uncultivated bacterium. At the time, the prevailing notion was that soil bacteria must have large genomes with diverse metabolisms to succeed. Instead, this species possesses a small genome and relies on high-affinity transporters to acquire nutrients directly from the environment. This work demonstrated that environmentally-dependent metabolisms can be advantageous and hinted at why so many soil bacteria resist culture.
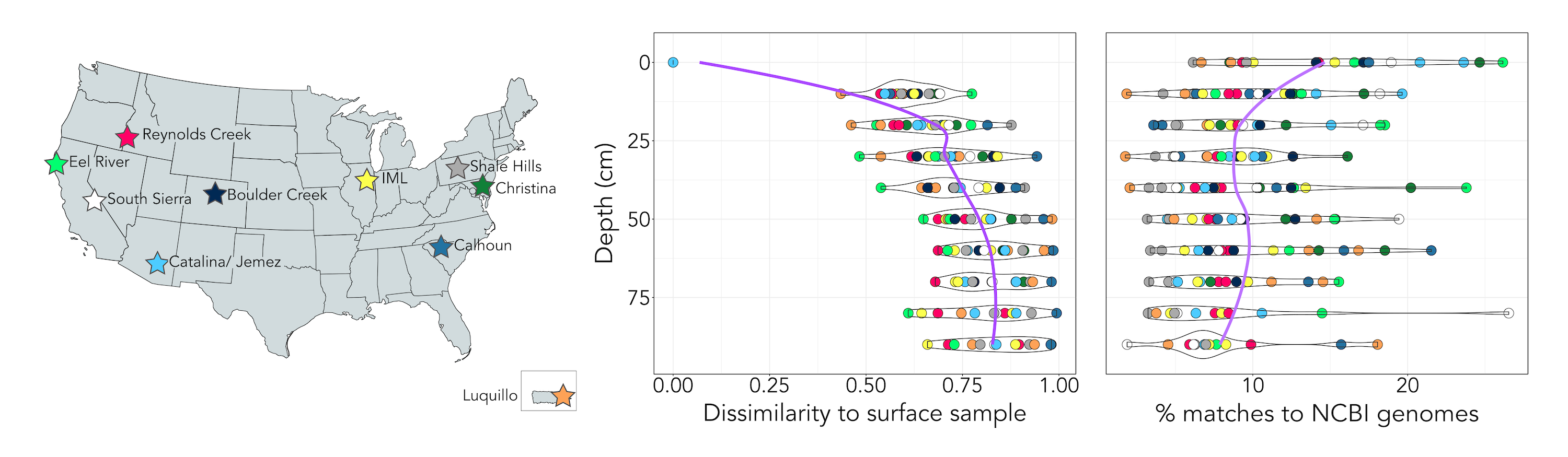
Deep soils harbor rare bacteria adapted to nutrient scarcity. This was a large collaborative project investigating soil microbial community changes with depth across the US. I found that deep soils exhibited unique communities underrepresented in genomic databases. I assembled and analyzed genomes from a candidate phylum which is particularly abundant in deep soils, the Dormibacterota. This phylum encodes adaptations for survival during periods of scarcity, including spore formation, ability to use carbon monoxide as an energy source, and the synthesis and storage of carbohydrates.
Methodological biases in microbial community sequencing techniques
Unlinked rRNA genes are widespread among bacteria and archaea. I found that many highly abundant, free-living species contain “unlinked” rRNA genes, where the 16S and 23S are separated by large chunks of the genome. This rearrangement is common in some environments—up to 41% of bacterial and archaeal rRNA genes are unlinked in soil. This work had a significant practical impact; the ubiquity of unlinked rRNA genes means that methods which rely on full rRNA operon genotyping (popular for resolving strain level differences), will fail to detect many species.
Extracellular, or relic DNA obscures temporal patterns in microbial communities. Here, I helped prove that extracellular DNA, or ‘relic DNA’, can obscure the true composition of microbial communities. Here, we used a DNA-binding dye (PMA) to prevent DNA from dead cells in soils from being sequenced. Once this relic DNA is removed, temporal patterns linked to changes in soil conditions are more readily observable. Together, both studies addressed methodological biases that can hinder a comprehensive understanding of microbial communities.